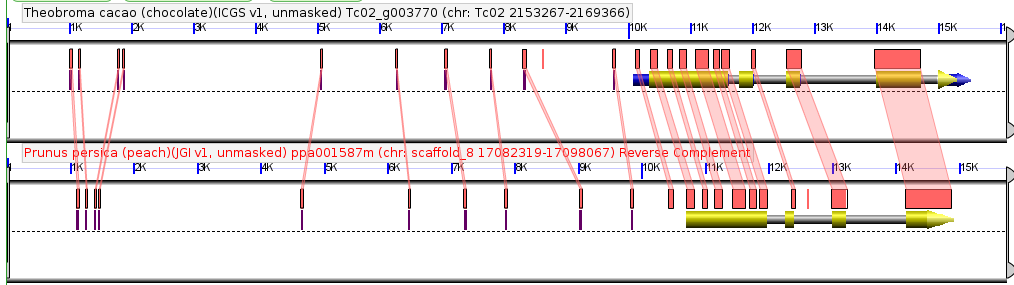
Inflorescence of Dichanthelium oligosanthes. Accession “Kellogg 1175”
Out of the ~12,000 known grass species, the genomes of less than one in one thousand have been sequenced. The “One in a Thousand” series focuses on these rare grass species.
Dichanthelium oligosanthes is a wild grass that grows in forest glades throughout the American midwest. It is a small plant. Doesn’t grow particularly fast. Its flowers aren’t particularly striking. And it has enough issues with seed dormancy that growing it in captivity is a major pain. Dichanthelium is a one in one-thousand grass with a sequenced reference genome.*
The reason folks are interested in Dichanthelium isn’t because of what it is, but who it’s related to. Dichanthelium occupies a spot on the grass family tree between a tribe** of grasses that includes foxtail millet and switchgrass, each one in a thousand species themselves, and another tribe of grasses that includes corn and sorghum, two more one in a thousand species. The relationship looks something like this:
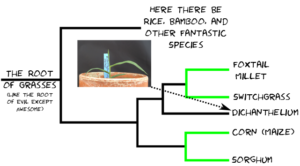
Phylogenetic relationship of Dichanthelium oligosanthes to related grasses with sequenced genomes.